
ARAVA
LEFLUNOMIDA
Tabletas
Frasco , 30 Tabletas recubiertas , 20 Miligramos
Caja , Blíster , 3 Tabletas , 100 Miligramos
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN
Ingrediente activo
Cada TABLETA de ARAVA® 20 mg contiene como ingrediente activo, 20 mg de leflunomida.
Cada TABLETA de ARAVA® 100 mg contiene como ingrediente activo, 100 mg de leflunomida.
Excipientes: C.s.
INDICACIONES: ARAVA® es usado para el tratamiento de la artritis reumatoide activa, para reducir los signos y síntomas, para inhibir la destrucción articular y para mejorar la función física y salud relacionados con la calidad de vida.
ARAVA® es usado para el tratamiento de Artritis psoriásica activa.
Si el paciente ya está recibiendo AINEs y/o corticosteroides en dosis bajas, estos pueden continuarse después de iniciar leflunomida.
El uso de leflunomida con antimaláricos usados en enfermedades reumáticas (p. ej., cloroquina e hidroxicloroquina), sales de oro intramusculares u orales, D-penicilamina, azatioprina y otros medicamentos inmunosupresores (p. ej., ciclosporina, metotrexato), no han sido adecuadamente estudiados.
FARMACOCINÉTICA: Leflunomida es convertida rápidamente al metabolito activo A771726, mediante metabolismo de primer paso (apertura del anillo) en la pared intestinal y el hígado. En un estudio con Leflunomida radio marcada con 14C en tres voluntarios sanos, no se detectó Leflunomida sin cambio en plasma, orina o heces. En otros estudios, los niveles de Leflunomida inmodificada en plasma, medidos en ng/mL rara vez han sido detectados. El único metabolito radio marcado detectado en plasma fue A771726. Este metabolito es el responsable de toda la actividad in vivo de ARAVA®.
Absorción: Los datos de excreción del estudio con 14C indicaron que al menos cerca del 82 al 95% de la dosis es absorbida. El tiempo para las concentraciones pico en plasma de A771726 es muy variable; los niveles pico en plasma pueden presentarse entre una hora y las 24 horas posteriores a la administración única.
Leflunomida puede ser administrada con alimentos, pues el grado de absorción es comparable con alimentos y en ayunas. Debido a la vida media larga de A771726, aproximadamente 2 semanas, una dosis de carga de 100 mg durante 3 días fue usada en los estudios, para lograr rápidas concentraciones plasmáticas del estado estable de A771726. Sin una dosis de carga, se estima que las concentraciones plasmáticas estables de A771726 se obtienen después de dos meses de dosificación. En estudios de dosis múltiples en pacientes con artritis reumatoide, los parámetros farmacocinéticos de A771726 fueron lineales a lo largo del rango de dosis de 5 a 25 mg. En estos estudios, el efecto clínico estuvo estrechamente relacionado con la concentración plasmática de A771726 y la dosis diaria de Leflunomida. Con un nivel de dosis de 20 mg/día, la concentración promedio en plasma de A771726 en estado estable es de aproximadamente 35 µg/mL. En estado estable los niveles en plasma se acumulan cerca de 33 a 35 veces, en comparación con la dosis única.
Distribución: En el plasma humano, A771726 se liga ampliamente a la proteína (albúmina). La fracción no ligada de A771726 es aproximadamente de 0,62%. El enlace de A771726 es lineal en el rango de concentración terapéutica. El enlace de A771726 aparece ligeramente reducido y más variable en el plasma de los pacientes con artritis reumatoide o insuficiencia renal crónica. El ligamiento extenso con la proteína del A771726 puede llevar a un desplazamiento de otros fármacos altamente ligados. Sin embargo, los estudios in vitro de interacción de enlace proteínico en plasma con warfarina en concentraciones clínicamente relevantes, no mostraron interacción. Estudios similares mostraron que ibuprofeno y diclofenaco no desplazan al A771726, mientras que la fracción no ligada de A771726 se aumenta en 2 a 3 veces en presencia de tolbutamida. A771726 desplazó al ibuprofeno, al diclofenaco y a la tolbutamida pero la fracción no ligada de estos medicamentos sólo se aumentó entre el 10 y 50%. No hay indicación de que estos efectos sean de relevancia clínica. En forma consistente con el enlace proteínico extenso, A771726 tiene un aparente bajo volumen de distribución (aproximadamente 11 L). No hay afinidad por los eritrocitos.
Metabolismo: Leflunomida es metabolizada a un metabolito principal (A771726) y otros muchos metabolitos menores incluyendo TFMA (4-trifluorometilanilina). La biotransformación metabólica de Leflunomida a A771726 y el metabolismo subsiguiente de A771726 no es controlado por una enzima única y ha sido demostrado que ocurre en las fracciones celulares microsómicas y citosólicas. Los estudios de interacción con cimetidina (inhibidor no específico de la citocromo P-450) y rifampicina (inductor no específico de la citocromo P-450) indican que las enzimas CYP in vivo están comprometidas en el metabolismo de Leflunomida sólo en grado mínimo.
Eliminación: La eliminación de A771726 es lenta y caracterizada por una depuración aparente de cerca de 31 mL/h. La vida media de eliminación en los pacientes es aproximadamente de 2 semanas. Después de la administración de una dosis radio marcada de Leflunomida, la radioactividad es excretada por igual en heces, probablemente por eliminación biliar, y orina. El A771726 es detectable en orina y en heces aún 36 días después de una administración única. Los principales metabolitos urinarios fueron los productos glucurónidos derivados de Leflunomida (principalmente en muestras de 0 a 24 horas) y un derivado del ácido oxanílico de A771726. El principal componente fecal fue A771726.
Se ha demostrado que en el hombre, la administración de una suspensión oral de carbón activado en polvo o de colestiramina, conduce a un aumento rápido y significativo en la tasa de eliminación de A771726 y una disminución en las concentraciones plasmáticas (ver sección Manejo de sobredosis). Se piensa que esto se logra gracias a un mecanismo de diálisis gastrointestinal y/o por la interrupción de la recirculación enterohepática.
• Poblaciones especiales
Insuficiencia renal: Leflunomida fue administrada como una dosis oral única de 100 mg a 3 pacientes con hemodiálisis y a 3 pacientes con diálisis peritoneal continua (CAPD). La farmacocinética del metabolito activo A771726 en los sujetos con CAPD fue similar a la de los voluntarios sanos. En los sujetos con hemodiálisis, ocurrió una eliminación más rápida del A771726, lo cual no fue debido a la extracción del fármaco por la diálisis sino al desplazamiento de la unión a las proteínas. El análisis de la cinética de 6 de esos pacientes demostró que, aunque la depuración del A771726 incrementó 2 veces la vida media de eliminación terminal, es similar a la de los sujetos sanos, dado que el volumen de distribución está también aumentado.
Insuficiencia hepática: No hay datos disponibles con respecto al tratamiento de los pacientes con deterioro hepático. El metabolito activo A771726 presenta una extensa unión a proteínas y es depurado por medio del metabolismo hepático y secreción biliar. Estos procesos pueden ser afectados por disfunción hepática.
Influencia de la edad: La farmacocinética en los niños y en los adolescentes no ha sido estudiada. Los datos farmacocinéticos en ancianos (>65 años de edad) son limitados pero consistentes con la farmacocinética en los adultos jóvenes.
Tabaquismo: Los datos de un análisis farmacocinético basado en la población de los estudios fase III indicaron que los fumadores tuvieron un incremento del 38% en la depuración sobre los no fumadores; sin embargo, no se vio ninguna diferencia en la eficacia clínica entre unos y otros.
FARMACODINAMIA
Farmacología humana: Leflunomida es un agente antirreumático modificador de la enfermedad con propiedades antiproliferativas. Leflunomida ha demostrado mejorar los signos y síntomas y desacelerar la progresión de la destrucción articular en la artritis reumatoide activa. En los respectivos estudios, la gran mayoría de pacientes usó concomitantemente AINEs o dosis bajas de corticosteroides.
Farmacología animal: Leflunomida es efectiva en modelos animales de artritis y de otras enfermedades autoinmunes y transplantes. Posee características inmunomoduladoras/inmunosupresoras, actúa como un agente antiproliferativo y exhibe propiedades antiinflamatorias.
In vivo, es metabolizado rápidamente y casi por completo a A771726, el cual es activo in vitro y es presumiblemente el producto medicinal activo.
Leflunomida presenta los mejores efectos protectores en modelos animales de enfermedades autoinmunes cuando se administra en la fase temprana de la progresión de la enfermedad. En modelos animales de rechazo crónico injerto contra huésped, y en rechazo de injerto de órgano sólido, Leflunomida ha prolongado el tiempo de rechazo o ha dado marcha atrás a las reacciones de rechazo en curso. Adicionalmente, Leflunomida presenta actividad antinflamatoria, aunque sólo una débil o nula actividad analgésica o antipirética. En un modelo de septicemia experimental, Leflunomida no alteró la resistencia de los ratones a los patógenos bacterianos.
Mecanismo de acción: A771726, el metabolito activo de Leflunomida, retarda el progreso de las células objetivo a través de diferentes fases del ciclo celular.
In vitro, después de la estimulación mitogénica, A771726, el metabolito activo, inhibe la proliferación de células T y la síntesis del DNA. Este inhibe la proliferación de las células mononucleares periféricas por estimulación mitogénica (PBMC) y la proliferación en las líneas celulares murinas y humanas transformadas, en forma dosis dependiente. Esta actividad antiproliferativa es reversada por la adición de uridina a los cultivos celulares, indicando que A771726 actúa a nivel de la biosíntesis de pirimidinas. Estudios de ligamento usando ligandos radiomarcados demuestran que A771726 se enlaza a la enzima dihidro-orotato-deshidrogenasa (DHODH) y la inhibe. Estos datos, en conjunto sugieren que, in vivo, en concentraciones alcanzables en pacientes que reciben Leflunomida puede inhibirse la síntesis de pirimidina en linfocitos y en otras poblaciones celulares de rápida división. La inhibición de la actividad de la tirosina-quinasa, tanto en situaciones in vitro como in vivo ha sido reportada. La actividad in vitro no parece ser mediada directamente a través de la inhibición enzimática y sólo tiene lugar en concentraciones mucho mayores de A771726 que las necesarias para la inhibición de la DHODH.
CONTRAINDICACIONES
ARAVA® no debe ser usado en: Pacientes con hipersensibilidad a Leflunomida, teriflunomida o a cualquiera de sus excipientes.
Mujeres embarazadas o mujeres fértiles que no estén usando un método de contracepción confiable durante el tratamiento con Leflunomida y mientras que los niveles en plasma del metabolito activo A 771726, estén sobre 0,02 mg/l (ver sección Embarazo). El embarazo debe ser descartado antes del inicio del tratamiento con Leflunomida.
La leflunomida no es recomendada en pacientes con daño hepático significativo ó con enfermedad hepática pre-existente y en infecciones serias activas.
EMBARAZO
Categoría de riesgo en el embarazo: X
No hay estudios clínicos que evalúen el uso de Leflunomida en mujeres embarazadas. Sin embargo, A771726 es teratogénico en ratas y conejos, y puede causar daño fetal en humanos.
Leflunomida está contraindicada en mujeres embarazadas o en mujeres potencialmente fértiles que no estén usando anticoncepción confiable (ver sección Interacciones) durante el tratamiento con Leflunomida y siempre que los niveles plasmáticos del metabolito activo A771726 estén por encima de 0,02 mg/L. El embarazo debe ser descartado antes de iniciar el tratamiento con Leflunomida.
Las pacientes deben ser advertidas de notificar inmediatamente al médico sobre cualquier retraso en la menstruación o cualquier otra razón para sospechar embarazo, para efectuar una prueba de embarazo y si es positiva, el médico y la paciente deben discutir el riesgo del embarazo. Es posible lograr un rápido descenso en los niveles sanguíneos del metabolito activo, mediante la implementación del procedimiento de eliminación del fármaco que se describe abajo, inmediatamente se detecte retraso en la menstruación, con el fin de disminuir el riesgo causado por Leflunomida en el feto.
Para las mujeres en tratamiento con Leflunomida que desean quedar embarazadas se recomienda uno de los siguientes procedimientos
— Después de suspender el tratamiento con Leflunomida, debe administrase 8 g. de colestiramina tres veces al día, por 11 días.
— Después de terminar el tratamiento con Leflunomida, debe administrarse 50 g. de carbón activado, 4 veces al día durante un periodo de 11 días.
Los 11 días no requieren ser consecutivos, a menos que haya necesidad de bajar rápidamente el nivel plasmático de A771726.
En cualquier caso, los niveles plasmáticos de A771726 < 0,02 mg/L deben ser verificados mediante dos pruebas separadas de por lo menos 14 días de diferencia. Se espera que niveles en plasma humano del metabolito activo menores a 0.02 mg/L (0.02 µg/mL) tengan un riesgo mínimo, con base en los datos disponibles.
Sin el procedimiento de eliminación, puede tomar hasta 2 años alcanzar los niveles de A771726 < 0,02 mg/L, debido a la variación individual en la depuración del medicamento. Sin embargo, incluso después de un periodo de espera de tal magnitud, la verificación de los niveles plasmáticos de A771726 < 0,02 mg/L a través de 2 mediciones independientes y separadas mínimo 14 días también es requerida.
No es posible garantizar una contracepción confiable con anticonceptivos orales durante el procedimiento de lavado con colestiramina o carbón activado. Se recomienda el uso de métodos anticonceptivos alternativos.
El riesgo de defectos al nacer y otras consecuencias adversas en el embarazo, que ocurran en mujeres que inadvertidamente quedan embarazadas mientras están en tratamiento con Leflunomida por un largo tiempo en el primer trimestre de embarazo son descritos en la sección Farmacodinamia.
LACTANCIA: Los estudios en animales indican que Leflunomida o sus metabolitos pasan a la leche materna. Sin embargo no se conoce si Leflunomida o sus metabolitos son excretados en la leche humana. Las mujeres, en consecuencia, no deben lactar mientras están recibiendo Leflunomida. La decisión deberá ser tomada con base en la necesidad de lactancia materna o la de iniciar tratamiento con Leflunomida, tomando en cuenta la importancia del medicamento para la madre.
CONDUCIR VEHÍCULOS O REALIZAR OTRAS TAREAS PELIGROSAS: No hay información disponible.
REACCIONES ADVERSAS
La siguiente frecuencia de puntuación CIOMS es usada cuando aplique: Muy Común (=10%), Común (=1% y <10%), Poco Común (=0.1% y <1%), Raro (=0.01 y <0.1%), Muy Raro (<0.01%). No conocido (no puede estimarse a partir de datos disponibles).
• Sistema gastrointestinal, hígado
Comunes: Diarrea, náusea, vómito, anorexia, desórdenes de la mucosa oral (como, p. ej., estomatitis aftosa, ulceraciones bucales), dolor abdominal, elevación de los parámetros hepáticos (como, p. ej., las transaminasas, menos frecuentemente gamma-GT, fosfatasa alcalina, bilirrubina), colitis, incluyendo colitis microscópica.
Raros: Hepatitis, ictericia/colestasis.
Muy raros: Daño hepático severo como falla hepática y necrosis hepática aguda que en algunos casos puede ser fatal. Pancreatitis.
• Sistema cardiovascular
Comunes: Aumento en la presión arterial.
No conocido: Hipertensión pulmonar.
• Sistema hemático y linfático
Comunes: Leucopenia con conteo de leucocitos > 2 x 109/L (> 2 G/L).
No comunes: Anemia, trombocitopenia con conteo de plaquetas < 100 x 109/L (<100 G/L).
Raros: Leucopenia con conteo de leucocitos < 2 x 109/L (<2 G/L), eosinofilia. Pancitopenia.
Recientemente, el uso concomitante o consecutivo de agentes potencialmente mielotóxicos puede estar asociado con un alto riesgo de efectos hematológicos.
• Sistema nervioso
Comunes: Cefalea, vértigo, parestesias.
No comunes: Desórdenes del gusto, ansiedad.
Muy raros: Neuropatía periférica.
• Reacciones alérgicas, piel y apéndices
Comunes: Reacciones alérgicas leves (incluyendo brote máculo-papular y otras erupciones), prurito, eczema, piel seca, incremento de la caída del cabello.
No comunes: Urticaria.
Muy raros: Reacciones anafilácticas/anafilactoides severas.
Síndrome de Stevens-Johnson (eritema multiforme de tipo mayor), necrólisis epidérmica tóxica. En los reportes de caso recibidos hasta ahora, no ha podido establecerse una relación causal con el tratamiento con Leflunomida, pero tampoco puede ser excluida.
Muy raro: Vasculitis, incluyendo vasculitis cutánea necrotizante.
Debido a la enfermedad subyacente, una relación causal no ha podido ser establecida.
• Infección
Raro: Infecciones severas y sepsis, las cuales pueden ser fatales.
La mayoría de los reportes de caso tuvieron como factor de confusión terapias inmunosupresoras concomitantes y/o enfermedad co-morbida adicional a la enfermedad reumatoide, lo cual puede predisponer a los pacientes a una infección.
Medicaciones como Leflunomida, la cual tiene potencial inmunosupresor, puede aumentar la susceptibilidad de los pacientes a la infección, incluyendo infecciones oportunistas (ver también la sección Precauciones).
En estudios clínicos, la incidencia por ejemplo de rinitis y bronquitis (5% vs. 2%), y neumonía (3% vs. 0%) estuvo levemente incrementada en los pacientes tratados con Leflunomida comparado con placebo, mientras que la incidencia global de infecciones fue comparable.
• Trastornos torácicos, respiratorios y mediastinales
Raro: Enfermedad pulmonar intersticial (Incluyendo neumonitis intersticial), la cual puede ser fatal.
• Trastornos de la piel y del tejido subcutáneo
Desconocido: Lupus eritematosos cutáneos, psoriasis pustular o empeoramiento de la psoriasis. Reacción del fármaco con eosinofilia y síntomas sistémicos (DRESS). Ver sección Precauciones.
• Otros
Comunes: Pérdida de peso, astenia.
No común: Hipocalemia.
Puede ocurrir hiperlipidemia leve. Los niveles de ácido úrico usualmente descienden debido a un efecto uricosúrico. Los posibles hallazgos adicionales de laboratorio para los cuales no ha podido establecerse relevancia clínica incluyen: Pequeños incrementos en LDH, CK y una pequeña disminución en los fosfatos.
Tendosinovitis y ruptura de tendón han sido reportados como eventos adversos en pacientes en tratamiento con Leflunomida, sin embargo una relación causal no ha podido ser establecida.
Disminuciones marginales (reversibles) en la concentración del esperma, conteo total espermático y motilidad rápida progresiva no pueden ser excluidas.
El riesgo de malignidad, en particular de desórdenes linfoproliferativos, se conoce que puede ser aumentado con el uso de algunos drogas inmunosupresoras.
INCOMPATIBILIDADES / COMPATIBILIDADES: No aplica.
INTERACCIONES: Un incremento en los efectos secundarios puede ocurrir en caso de uso reciente o concomitante de sustancias hepatotóxicas (incluye alcohol), hematotóxicas o inmunosupresoras. Esto debe ser considerado cuando el tratamiento con Leflunomida sea seguido por tales sustancias sin un periodo previo de lavado. Ver también sección Precauciones.
Metotrexato: En un estudio pequeño (n=30) en pacientes con Artritis Reumatoidea (AR) en el cual se co-administró leflunomida (10 a 20 mg por día) con metotrexato (10 a 25 mg por semana) una elevación de 2 a 3 veces el nivel de las enzimas hepáticas fue observado en 5 de los 30 pacientes. Todas las elevaciones se resolvieron, 2 con la continuación de ambos fármacos y 3 después de la suspensión de Leflunomida. Un incremento mayor de 3 veces los niveles basales fue observado en otros 5 pacientes. Todos esos casos también se resolvieron, 2 con continuación de ambos medicamentos y 3 después de la suspensión de Leflunomida. En consecuencia, aunque, en general, no es necesario un periodo de espera cuando se cambia de Leflunomida a metotrexato, un monitoreo estrecho de las enzimas hepáticas es recomendado en la fase inicial después del cambio.
Vacunas: No se dispone de datos clínicos sobre la eficacia y seguridad de las vacunas durante el tratamiento con Leflunomida. Sin embargo, no se recomienda la vacunación con vacunas vivas. La larga vida media de la Leflunomida debe considerarse cuando se contempla la administración de una vacuna viva después de la suspensión de Leflunomida.
Warfarina: Ha habido casos reportados de aumento del tiempo de protrombina, cuando leflunomida y la warfarina se administraron conjuntamente. Una interacción farmacodinámica con Warfarina se observó con A771726 en un estudio farmacológico clínico (ver más abajo). Por lo tanto, cuando la Warfarina se co-administra, se recomienda el estrecho seguimiento y monitoreo con INR.
Alimentos: El grado de absorción de leflunomida no se ve afectada cuando se toma con alimentos.
Efecto de otros fármacos sobre leflunomida: En estudios de inhibición in vitro en microsomas de hígado humano sugieren que el citocromo P450 (CYP) 1A2, 2C19 y 3A4 están involucrados en el metabolismo de Leflunomida. En un estudio de interacción in vivo con Leflunomida y cimetidina (inhibidor leve no específico de la citocromo P-450 (CYP)) se ha demostrado falta de un impacto significativo en la exposición de A771726.
Posterior a la administración concomitante de una dosis única de Leflunomida a sujetos que recibiendo dosis múltiples de rifampicina (inductor no específico de la citocromo P-450) los niveles pico de A771726 se incrementaron aproximadamente 40%, mientras que el área bajo la curva (AUC) no cambió significativamente. El mecanismo de este efecto no es claro. El potencial para continuar el incremento de los niveles de Leflunomida con dosificación múltiple puede necesitar consideración adicional si los pacientes están recibiendo concomitantemente Leflunomida y rifampicina.
La administración de colestiramina o carbón activado conduce a una disminución rápida y significativa en la concentración plasmática de A771726. El mecanismo se cree que es por la interrupción del ciclo enterohepático y / o diálisis gastrointestinal del A771726. Ver también las secciones Embarazo y Sobredosis.
Efecto de la leflunomida sobre otros fármacos
— Sustratos BCRP: Aunque una interacción farmacocinética con un sustrato de BCRP (rosuvastatina) se observó con el A771726 (ver más abajo), fue demostrado que no hay interacción farmacocinética, en 12 pacientes, entre leflunomida (10 a 20 mg por día) y metotrexato (un sustrato de BCRP; 10 a 25 mg por semana).
— Los estudios de interacción medicamentosa in vivo han demostrado que no existe interacción medicamentosa significativa entre Leflunomida y los anticonceptivos trifásicos orales. En un estudio en el cual Leflunomida fue administrada concomitantemente con una píldora anticonceptiva trifásica oral que contenía 30 µg de etinil-estradiol a mujeres voluntarias saludables, no hubo reducción en la actividad contraceptiva de la píldora y la farmacocinética de A771726 estuvo dentro de los rangos predictivos. Una interacción farmacocinética con los anticonceptivos orales se observó con A771726 (ver abajo).
Se realizaron los siguientes estudios de interacción farmacocinética y farmacodinamica con A771726 (principal metabolito activo de la Leflunomida). Como interacciones similares entre fármacos no pueden ser excluidos por leflunomida en las dosis recomendadas, los siguientes resultados y recomendaciones del estudio deben considerarse en los pacientes tratados con leflunomida:
Efecto sobre la repaglinida (sustrato de CYP2C8): Hubo un aumento en el promedio de repaglinida Cmáx y área bajo la curva (AUC) (1,7 y 2,4 veces, respectivamente), después de dosis repetidas de A771726, lo que sugiere que A771726 es un inhibidor del CYP2C8 in vivo. Por lo tanto, el seguimiento de los pacientes con el uso concomitante de fármacos metabolizados por CYP2C8, como la repaglinida, paclitaxel, pioglitazona o rosiglitazona, se recomienda ya que pueden tener una mayor exposición.
Efecto de la cafeína (sustrato de CYP1A2): Las dosis repetidas de A771726 disminuyeron significativamente la Cmáx y el área bajo la curva (AUC) de cafeína (sustrato de CYP1A2) en un 18% y 55%, respectivamente, lo que sugiere que la A771726 puede ser un inductor débil de CYP1A2 in vivo. Por lo tanto, los medicamentos metabolizados por el CYP1A2 (p. ej., duloxetina, alosetron, teofilina y tizanidina) deben utilizarse con precaución durante el tratamiento concomitante, ya que podría llevar a la reducción de la eficacia de estos productos.
Efecto sobre el transportador de sustratos de aniones orgánicos 3 (OAT3): Hubo un aumento en la media de cefaclor Cmáx y AUC (1,43 y 1,54 veces, respectivamente), después de dosis repetidas de A771726, lo que sugiere que la A771726 es un inhibidor de la OAT3 in vivo. Por lo tanto, cuando se coadministra con sustratos de OAT3, tales como cefaclor, bencilpenicilina, ciprofloxacina, indometacina, ketoprofeno, furosemida, cimetidina, metotrexato, zidovudina, se recomienda precaución.
Efecto en el BCRP y/ o Substratos de péptidos transportadores de aniones orgánicos B1 / B3: Hubo un aumento en el promedio de la rosuvastatina Cmáx y AUC (2,65 y 2,51 veces, respectivamente), después de dosis repetidas de A771726. Sin embargo, no hubo un impacto aparente de este aumento en la exposición de rosuvastatina en plasma sobre la actividad de la HMG-CoA reductasa. Si se usan juntos, la dosis de rosuvastatina no debe exceder de 10 mg una vez al día. Para otros sustratos de BCRP (p. ej., metotrexato, topotecán, sulfasalazina, daunorrubicina, doxorrubicina) y la familia de OATP especialmente la de los inhibidores de la HMG-CoA reductasa (p. ej., simvastatina, atorvastatina, pravastatina, metotrexato, nateglinida, repaglinida, rifampicina) la administración concomitante debe también realizarse con precaución. Los pacientes deben ser estrechamente monitorizados para detectar signos y síntomas de una exposición excesiva a los medicamentos y por lo tanto la reducción de la dosis de estos medicamentos debe ser considerada.
Efecto sobre el anticonceptivo oral (etinilestradiol 0,03 mg y levonorgestrel 0,15 mg): Hubo un aumento en el promedio de etinilestradiol Cmáx y el AUC0-24 (1,58 y 1,54 veces, respectivamente) y levonorgestrel Cmáx y AUC0-24 (1,33 y 1,41 veces, respectivamente) tras repetidas dosis de A771726. Si bien no se espera que esta interacción impacte negativamente la eficacia de los anticonceptivos orales, se debe poner en consideración el tipo de tratamiento anticonceptivo oral.
Efecto sobre la Warfarina: Dosis repetidas de A771726 no tuvieron efecto sobre la farmacocinética de la S-Warfarina, lo que indica que A771726 no es un inhibidor o un inductor de CYP2C9. Sin embargo, se observó una disminución del 25% en el pico del índice internacional normalizado (INR) cuando se co-administra Warfarina con A771726 en comparación con la Warfarina sola. Por lo tanto, cuando la Warfarina se co-administra, se recomienda el estrecho seguimiento y monitoreo del INR.
DATOS DE SEGURIDAD NO CLÍNICA
Toxicidad aguda: Leflunomida, administrada oral e intraperitonealmente, ha sido analizada en estudios de toxicidad aguda en ratones y ratas.
En los ratones, los valores de LD50 oral fluctuaron entre 200 y 500 mg/Kg y en las ratas entre 100 y 250 mg/Kg. Posterior a la administración intraperitoneal, los valores LD50 fueron de aproximadamente 400 mg/Kg en los ratones y entre 200 y 400 mg/Kg en las ratas.
Toxicidad crónica: La administración oral repetida de Leflunomida a ratas y perros hasta por 6 meses, no reveló efectos de las dosis de 0,5 y 0,8 mg/Kg/día, respectivamente. Dosis mayores causaron cambios patológicos en ratas, en especial hipoplasia de la médula ósea, trombocitopoyesis esplénica reducida, atrofia del timo, hemorragias en el tracto gastrointestinal y otros tejidos, y muerte. Con dosis de 1 mg/Kg/día o mayores, se presentaron anemia y eritropoyesis extramedular esplénica. En perros se notó reducción de los parámetros de la eritropoyesis, presencia de cuerpos de Heinz y/o cuerpos de Howell-Jolly, hemopoyesis extramedular y hemosiderosis. Las muertes se presentaron en perros a los cuales se les administraron 8 mg/Kg/día.
Debido a su actividad farmacodinámica, leflunomida inhibe la proliferación y la diferenciación celular. Consecuentemente, los efectos sobre los órganos reproductores se vieron en estudios de dosis repetida en ratones, con niveles altos de medicamento (degeneración y atrofia de testículos, próstata, vesículas seminales, con 30 mg/Kg de peso corporal, y atrofia de ovarios y útero con 100 mg/Kg de peso corporal). En los perros, fue observada disminución de peso de la próstata y los testículos en el grupo de altas dosis (8 mg/Kg de peso corporal), estudio de 3 meses.
Mutagénesis: Leflunomida no fue mutagénica en el ensayo de Ames, ensayo no-programado de síntesis de DNA o en el ensayo de mutación genética HGPRT. Adicionalmente, Leflunomida no fue clastogénica en el ensayo in vivo de micronúcleos de ratón, ni en la prueba citogenética in vivo en células de médula ósea de hámster chino.
Sin embargo, la 4-trifluorometilanilina (TFMA), un metabolito menor de la Leflunomida, fue mutagénica en el ensayo Ames y en el ensayo de mutación genética HGPRT y clastogénica en el ensayo in vitro de aberraciones cromosómicas en células de hámster chino. La TFMA no fue clastogénica en el ensayo in vivo de micronúcleos de ratón, ni en la prueba citogénica in vivo en células de médula ósea de hámster chino.
Carcinogenicidad: No se observó evidencia alguna de carcinogenicidad en un bio-ensayo de 2 años en ratas con dosis orales de Leflunomida hasta dosis máxima de 6 mg/Kg (aproximadamente 1/40 de la exposición sistémica humana máxima M1 con base en la AUC). Sin embargo, ratones machos en un bio-ensayo de 2 años presentaron una incidencia mayor de linfoma con una dosis oral de 15 mg/Kg, la dosis más alta estudiada (1,7 veces la exposición humana M1 basada en la AUC). En el mismo estudio, los ratones hembras presentaron una mayor incidencia relacionada con la dosis de adenomas y carcinomas bronco alveolares combinados, empezando en 1,5 mg/Kg (aproximadamente 1/10 de la exposición humana M1 basada en la AUC). No se conoce el significado de los hallazgos en ratones relativo al uso clínico de Leflunomida.
Antigenicidad: Leflunomida no fue antigénica en la prueba de anafilaxis cutánea pasiva y sistémica activa, llevada a cabo en cobayos y estuvo exenta de propiedades sensibilizantes.
Teratogenicidad: A771726 es teratogénico en ratas y conejos.
Leflunomida, cuando fue administrada oralmente a ratas durante la organogénesis en una dosis de 15 mg/Kg, fue teratogénica (siendo lo más notable anoftalmia o microftalmia e hidrocéfalo interno). La exposición sistémica de ratas en esta dosis fue de aproximadamente 1/10 del nivel de la exposición humana basada en la AUC. Bajo estas condiciones de exposición, Leflunomida causó un descenso en el peso corporal materno y un aumento en la embrio-letalidad, con un descenso en el peso corporal fetal en los fetos sobrevivientes. En los conejos, el tratamiento oral con 10 mg/Kg de Leflunomida durante la organogénesis tuvo como resultado fusión y esternebras displásicas. El nivel de exposición con esta dosis fue en esencia equivalente al máximo nivel de exposición humana con base en la AUC. En una dosis de 1 mg/Kg, Leflunomida no fue teratogénica en ratas y conejos.
Cuando las ratas hembras fueron tratadas con 1,25 mg/Kg de Leflunomida, empezando 14 días antes del apareamiento y continuando hasta el fin de la lactancia, la descendencia presentó descensos notables (mayores al 90%) en la sobrevida postnatal. El nivel de exposición sistémica con 1,25 mg/Kg fue de aproximadamente 1/100 del nivel de exposición humana con base en la AUC.
EFICACIA CLÍNICA
Artritis reumatoidea: La eficacia de Leflunomida en el tratamiento de la artritis reumatoidea (RA) se demostró en tres estudios controlados en los cuales se evidenció la reducción en signos y síntomas, y la inhibición del daño estructural. En dos de los tres estudios controlados, también se demostró la mejoría de la función física.
En todos los estudios de monoterapia con Leflunomida sólo se usó una dosis inicial de carga de 100 mg al día, durante tres días, seguida de 20 mg al día, en lo sucesivo.
US301: En el estudio US301, un estudio controlado con placebo de 2 años de duración, se asignaron al azar 482 pacientes con AR activa de por lo menos 6 meses de duración a recibir leflunomida 20 mg/día (n=182), metotrexato 7.5 mg/semana aumentando hasta 15 mg/semana (n=182), o placebo (n=118). Todos los pacientes recibieron folato 1 mg BID. El análisis primario se hizo a las 52 semanas y el tratamiento continuó a ciegas hasta las 104 semanas.
En general, 235 de los 508 pacientes tratados asignados al azar (482 en el análisis primario de datos y otros 26 pacientes adicionales), continuaron en un segundo tratamiento doble-ciego de 12 meses de duración (98 leflunomida, 101 metotrexato, 36 placebo). Las dosis de leflunomida continuó a 20 mg/día y la dosis de metotrexato se pudo aumentar hasta un máximo de 20 mg/semana. En total 190 pacientes (83 leflunomida, 80 metotrexato, 27 placebo) completaron 2 años de tratamiento doble-ciego.
MN301/303/305: En el estudio MN301, un estudio controlado con placebo, se asignaron al azar 358 pacientes con AR activa a recibir leflunomida 20 mg/día (n=133), sulfasalazina 2.0 g/día (n=133), o placebo (n=92).
La duración del tratamiento fue de 24 semanas. Una extensión del estudio fue una continuación opcional de 6 meses de duración, controlada a ciegas con sustancia activa y no con placebo, MN301, la cual dio lugar a una comparación de 12 meses de leflunomida y sulfasalazina (estudio MN303).
De los 168 pacientes que completaron 12 meses de tratamiento en el MN301 y el MN303, 146 pacientes (87%) entraron en un estudio de extensión de 1 año de tratamiento activo, doble-ciego controlado, con sustancia activa y no con placebo (MN305; 60 leflunomida, 60 sulfasalazina, 26 placebo/ sulfasalazina). Los pacientes continuaron con la misma dosis diaria de leflunomida o sulfasalazina que estaban tomando cuando terminaron el MN301/303. En total 121 pacientes (53 leflunomida, 47 sulfasalazina, 21 placebo / sulfasalazina) completaron los 2 años de tratamiento doble-ciego.
MN302/304: En el estudio MN302, un estudio controlado con sustancia activa y no con placebo, se asignaron al azar 999 pacientes con AR activa a recibir leflunomida 20 mg/día (n=501) o metotrexato en dosis de 7.5 mg/semana aumentando hasta 15 mg/semana (n=498). Se utilizó suplementación con folato en 10% de los pacientes. La duración del tratamiento fue de 52 semanas.
De los 736 pacientes que completaron las 52 semanas de tratamiento en el estudio MN302, 612 (83%) entraron en el estudio de extensión doble-ciego, de un año de duración identificado como MN304 (292 leflunomida, 320 metotrexato). Los pacientes continuaron tomando la misma dosis diaria de leflunomida o metotrexato que habían estado tomando cuando completaron el MN302. Hubo 533 pacientes (256 leflunomida, 277 metotrexato) que completaron 2 años de tratamiento doble-ciego.
Signos y síntomas de la artritis reumatoidea: El alivio de los signos y síntomas se valoró usando el Indice de Respuesta del Colegio Americano de Reumatología (ACR 20), un índice compuesto de medidas clínicas, de laboratorio y funcionales en artritis reumatoidea. Un paciente “ACR20 respondedor” es aquel que tuvo una mejora >20% en el conteo de articulaciones inflamadas y tumefactas y en 3 de los siguientes 5 criterios: Valoración global del médico, valoración global del paciente, medición de capacidad funcional [Cuestionario Modificado de Valoración Médica (MHAQ por sus siglas en inglés)], escala visual análoga de dolor, y velocidad de sedimentación globular o proteína C reactiva. Un paciente “ACR20 respondedor en el Punto Final” es aquel que completó el estudio y fue ACR20 respondedor cuando terminó el estudio.
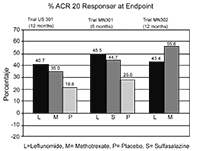
Las tasas de ACR20 respondedores en el Punto Final se muestran en la Figura 1. Leflunomida fue superior, en grado estadísticamente significativo al placebo, en la reducción de los signos y síntomas de AR según el análisis primario de eficacia, es decir, pacientes ACR20 Positivos en el Punto Final, en el estudio US301 (en el punto final primario de los 12 meses) y en el MN301 (en el punto final de los 6 meses). Las tasas de pacientes ACR20 respondedores en el Punto Final con Leflunomida fueron concordantes a través de los estudios de 6 y 12 meses (41 - 49%). No se demostraron diferencias constantes durante el tratamiento entre leflunomida y metotrexato o entre leflunomida y sulfasalazina. El efecto del tratamiento con Leflunomida se hizo evidente hacia el mes 1, se estabilizó hacia los 3 - 6 meses, y continuó durante todo el curso del tratamiento como se muestra en la Figura 1.
Figura 1
|
Comparaciones |
Intervalo Confianza 95% |
Valor p |
|
|
US301 |
Leflunomida vs. Placebo |
(12, 32) |
<0,0001 |
|
Metotrexato vs. Placebo |
(8, 30) |
<0,0001 |
|
|
Leflunomida vs. Metotrexato |
(-4, 16) |
NS |
|
|
MN301 |
Leflunomida vs. Placebo |
(7, 33) |
0,0026 |
|
Sulfasalazina vs. Placebo |
(4, 29) |
0,0121 |
|
|
Leflunomida vs. Sulfasalazina |
(-8, 16) |
NS |
|
|
MN302 |
Leflunomida vs. Metotrexato |
(-19,-7) |
<0,0001 |
Figura 2
Pacientes ACR Positivos del US301
en el curso del tiempo
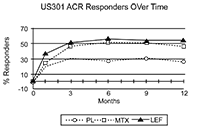
*Ultima observación llevada a cabo.
Los pacientes ACR50 y ACR70 Positivos se definen de manera análoga a los respondedores ACR 20, pero para ellos se usan mejoras del 50% o 70%, respectivamente (tabla 1). El cambio promedio de los componentes individuales del Indice de respondedores ACR se muestra en la Tabla 1.
|
Tabla 1. Resumen de las tasas de Respuesta ACR* |
|||
|
Grupo de Estudio y Tratamiento |
ACR20 |
ACR50 |
ACR70 |
|
Estudios Controlados con Placebo |
|||
|
US301 (12meses) |
|
|
|
|
Leflunomida (n=178)+ |
52,2 ++ |
34,3++ |
20,2++ |
|
Placebo (n=118)+ |
26,3 |
7,6 |
4,2 |
|
Metotrexato (n=180)+ |
45,6 |
22,8 |
9,4 |
|
MN301 (6meses) |
|
|
|
|
Leflunomida (n=130)+ |
54,6++ |
33,1++ |
10,0& |
|
Placebo (n=91)+ |
28,6 |
14,3 |
2,2 |
|
Sulfasalazina (n=132)+ |
56,8 |
30,3 |
7,6 |
|
Estudios controlados con sustancia activa y no con placebo |
|||
|
MN302 (12meses) |
|
|
|
|
Leflunomida (n=495)+ |
51,1 |
31,1 |
9,9 |
|
Metotrexato (n=489)+ |
65,2 |
43,8 |
16,4 |
|
*Análisis de intención de tratar (IDT) usando la técnica de la última observación llevada a cabo (UOAA) para los pacientes que abandonaron antes de tiempo. +N es el número de pacientes de IDT para quienes los datos estuvieron disponibles para calcular las tasas indicadas. ++ p<0,001 leflunomida vs placebo & p>0,02 leflunomida vs placebo |
|||
La Tabla 2 muestra los resultados de los componentes de los criterios de respuesta ACR para los estudios US301, MN301, y MN302. Leflunomida fue significativamente superior al placebo en todos los componentes de los criterios de respuesta ACR en los estudios US301 y MN301. Además, la Leflunomida fue significativamente superior al placebo en la mejora de la rigidez matinal, una medida de la actividad de la enfermedad reumática, que no está incluida en los criterios de respuesta ACR No se demostraron diferencias constantes entre Leflunomida y los comparadores activos.
|
Tabla 2. Cambio promedio en comparación del índice de Respuesta ACR * |
||||||||
|
Componentes |
Estudios controlados con placebo |
Estudio Controlado con activo |
||||||
|
|
US301 |
MN301 No EU |
MN302 No EU |
|||||
|
|
(12 meses) |
(6 meses) |
(12 meses) |
|||||
|
Lef |
Met |
Pla |
Lef |
Sul |
Pla |
Lef |
Met |
|
|
Recuento del dolor articular,1 |
-7,7 |
-6,6 |
-3,0 |
-9,7 |
-8,1 |
-4,3 |
-8,3 |
-9,7 |
|
Recuento de Tumefacción,1 |
-5,7 |
-5,4 |
-2,9 |
-7,2 |
-6,2 |
-3,4 |
-6,8 |
-9,0 |
|
Valoración global el paciente 2 |
-2,1 |
-1,5 |
0,1 |
-2,8 |
-2,6 |
-0,9 |
-2,3 |
-3,0 |
|
Valoración global del médico 2 |
-2,8 |
-2,4 |
-1,0 |
-2,7 |
-2,5 |
-0,8 |
-2,3 |
-3,1 |
|
Función/discapacidad física (MHAQ/HAQ) |
-0,29 |
-0,15 |
0,07 |
-0,5 |
-0,29 |
-0,04 |
-0,37 |
-0,44 |
|
Intensidad del dolor 2 |
-2,2 |
-1,7 |
-0,5 |
-2,7 |
-2,0 |
-0,9 |
-2,1 |
-2,9 |
|
Velocidad de Eritrosedimentación |
-6,26 |
-6,48 |
2,56 |
-7,48 |
-16,56 |
3,44 |
-10,12 |
-22,18 |
|
Proteína C reactiva |
-0,62 |
-0,5 |
0,47 |
-2,26 |
-1,19 |
0,16 |
-1,86 |
-2,45 |
|
No incluido en el índice de Respuesta ACR |
||||||||
|
Rigidez matinal (min) |
-101,4 |
-88,7 |
14,7 |
-93 |
-42,4 |
-6,8 |
-63,7 |
-86,6 |
|
*Ultima observación llevada a cabo; Cambio negativo indica mejora. 1 Basado en el conteo de 28 articulaciones 2 Escala visual análoga 0=mejor; 10= peor |
||||||||
Mantenimiento del efecto: Después de completar 12 meses de tratamiento, los pacientes que siguieron tomando el tratamiento del estudio fueron evaluados durante otros 12 meses adicionales de tratamiento doble-ciego (periodo total de tratamiento de 2 años) en los estudios US301, MN305, y MN304. Las tasas de respuesta ACR positiva a los 12 meses se mantuvieron durante los 2 años en la mayoría de los pacientes que continuaron un segundo año de tratamiento.
La mejora desde la situación basal en los componentes individuales de los criterios de respondedores ACR también se mantuvo en la mayor parte de los pacientes durante el segundo año de tratamiento con Leflunomida en los tres estudios.
b. Inhibición del daño estructural
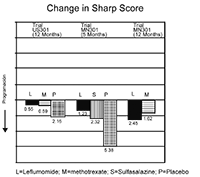
La inhibición del daño estructural comparado con el control se valoró usando la Puntuación de Sharp, una escala compuesta de erosiones evidenciadas por rayos x y estrechamiento del espacio articular en las manos, los puños y la parte anterior del pie. El cambio desde la situación basal hasta el punto final en la progresión de la enfermedad estructural, medida por la puntuación de rayos X de Sharp, se muestra en la Figura 3. ARAVA® fue estadísticamente superior al placebo, en la inhibición del avance de la enfermedad mediante la Puntuación de Sharp. No se demostraron diferencias relevantes durante el tratamiento entre leflunomida y metotrexato o entre leflunomida y sulfasalazina.
Figura 3
Cambio en la puntuación de Sharp
|
Comparaciones |
Intervalo de confianza 95% |
Valor p |
|
|
US301 |
Leflunomida vs. Placebo |
(-4,0, -1,1) |
0,0007 |
|
Metotrexato vs. Placebo |
(-2,6, -0,2) |
0,0196 |
|
|
Leflunomida vs. Metotrexato |
(-2,3, 0,0) |
0,0499 |
|
|
MN301 |
Leflunomida vs. Placebo |
(-6,2, -1,8) |
0,0004 |
|
Sulfasalazina vs. Placebo |
(-6,9, 0,0) |
0,0484 |
|
|
Leflunomida vs. Sulfasalazina |
(-3,3, 1,2) |
NS |
|
|
MN302 |
Leflunomida vs. Metotrexato |
(-2,2, 7,4) |
NS |
c. Mejora de la función física
La mejora de la función física se valoró usando el Cuestionario de Valoración de la Salud (HAQ) y el Formulario reducido de la Encuesta de Resultados Médicos (SF-36). El Cuestionario de Valoración de la Salud (HAQ) valora la función física del paciente y su grado de discapacidad. El cambio medio desde la situación basal en la capacidad funcional medido con el Índice de Discapacidad del HAQ (HAQ DI) en los estudios de 6 y 12 meses controlados con placebo y con sustancia activa se muestra en la Figura 4. La Leflunomida fue superior al placebo en grado estadísticamente significativo en la mejora de la función física. La superioridad al placebo se demostró de forma constante en las ocho escalas secundarias del HAQ DI (vestirse, incorporarse, comer, caminar, higiene, alcanzar, agarrar y actividades) en ambos estudios controlados con placebo.
El Formulario reducido de la Encuesta de Resultados Médicos 36 (SF-36), un cuestionario genérico de la calidad de vida relacionada con la salud, considera además la función física. En el estudio US301, a los 12 meses la leflunomida produjo mejoras estadísticamente significativas, comparada con el placebo en el Puntuación Resumida del Componente Físico (PCS).
Figura 4.
Cambio en la medición de la capacidad funcional*

|
Comparación |
Intervalo Confianza 95% |
Valor p |
|
|
US301 |
Leflunomida vs. Placebo |
(-0,58, -0,29) |
0,0001 |
|
Leflunomida vs. Metotrexato |
(-0,34, -0,07) |
0,0026 |
|
|
MN301 |
Leflunomida vs. Placebo |
(-0,67, -0,36) |
<0,0001 |
|
Leflunomida vs. Sulfasalazina |
(-0,33, -0,03) |
0,0163 |
|
|
MN302 |
Leflunomida vs. Metotrexato |
(0,01, 0,16) |
0.221 |
Mantenimiento del efecto: La mejora de la función física demostrada a los 6 y 12 meses se mantuvo durante dos años. En aquellos pacientes que continuaron terapia durante un segundo año, esta mejora de la función física medida con el HAQ y el SF-36 (PCS) se mantuvo.
Artritis psoriásica: Se asignaron al azar pacientes adultos con artritis psoriásica (APs) bien sea a recibir leflunomida o placebo. La duración del tratamiento fue de 6 meses y las dosis fueron de 100 mg/día durante tres días seguido de 20 mg/día de leflunomida durante el resto del periodo. En el grupo de leflunomida, de los pacientes que fueron completamente analizables (n=186), 59,0% tuvieron una mejora de los Criterios de Respuesta al Tratamiento de la Artritis Psoriásica (PsARC), el punto final primario, comparados con 29,7% en el grupo de placebo (p<0.0001). El PsARC por sus siglas en inglés, es una medida que combina la valoración global del médico, una evaluación de seguridad global del paciente, una puntuación de dolor / sensibilidad articular y una puntuación de tumefacción articular. La mejora de los PsARC se define como una disminución de > a 30% de las puntuaciones articulares y de un punto para las valoraciones globales. Una mejora de por lo menos 2 de los anteriores, uno de los cuales ha de ser la puntuación de dolor / sensibilidad o de tumefacción articular y no empeoramiento de ninguna de las 4 medidas se requirió para considerar al paciente como un respondedor. (Ver la Tabla 3).
|
Tabla 3: Cambio Medio en el Índice de Respuesta a los Componentes de los PsARC* |
|||
|
Componentes |
Estudio controlado con placebo (6 meses) |
||
|
Leflunomida (n=95) |
Placebo (n=91) |
Valores p |
|
|
Puntuación de dolor/ |
-9,1 |
-4,6 |
0,0022 |
|
Puntuación de tumefacción articular* |
-6,8 |
-4,2 |
0,0013 |
|
Valoración global del médico – mejora de por lo menos una categoría1 |
52,6% |
34,1% |
< 0,0001 |
|
Autovaloración global del paciente– mejora de por lo menos una categoría1 |
31,6% |
30.8% |
0.0036 |
|
*Cambio negativo indica mejora Escala de Likert de 1 a 5 puntos donde 1 = Muy bueno; 5 = Muy malo |
|||
Los cambios en el Indice de Area y Gravedad de la Psoriasis (PASI) reflejan cambios en la extensión y la gravedad de las lesiones de psoriasis a juzgar por eritema, descamación e infiltración. La Leflunomida dio lugar a una mejoría significativa en las puntuaciones PASI en el curso de las 24 semanas del estudio con respecto al placebo, con una mejora media (± DE) de 22,4% (± 51,6%) en el grupo de leflunomida comparada con un deterioro de 2,2% (± 70,4%) en el grupo de placebo (p = 0,0030). Comparado con el grupo de placebo, una proporción significativamente mayor de pacientes del grupo de leflunomida experimentó una reducción >50% en las puntuaciones PASI (PASI 50; 18.9% vs. 30.4%; p = 0.050) y una reducción de las puntuaciones PASI >75% (PASI 75; 7,8% vs. 17,4%; p = 0.048) desde la situación basal. Los eventos adversos observados en el estudio clínico en pacientes con Artritis Psoriásica fueron comparables con los eventos adversos vistos en los estudios clínicos en pacientes con artritis reumatoidea.
Exposición durante el embarazo: Leflunomida no debe ser empleada en mujeres embarazadas, o en mujeres que pueden quedar en embarazo si no están utilizando un método de contracepción confiable durante el tratamiento con leflunomida. El embarazo debe ser descartado antes del inicio del tratamiento con leflunomida. Se debe aconsejar a los pacientes que ante el riesgo o sospecha de embarazo, deben notificar inmediatamente a su médico tratante para realizar una prueba de embarazo y si ésta es positiva, se deben discutir los riesgos relacionados con el desenlace del embarazo. Los procedimientos para eliminación del fármaco para bajar rápidamente los niveles sanguíneos del metabolito activo pueden disminuir el riesgo para el feto (ver secciones Contraindicaciones y Embarazo).
Un estudio prospectivo de los desenlaces del embarazo para determinar el riesgo de defectos al nacimiento y otros desenlaces adversos debido a al consumo inadvertido de leflunomida durante el primer trimestre de embarazo, fue realizado por la Organización de Especialistas en Información sobre Teratología (OTIS, en inglés: Organization of Teratotology Information Specialists). Las mujeres en embarazo fueron reclutadas en uno de tres grupos: Mujeres con diagnóstico de artritis reumatoidea que tomaron al menos una dosis de leflunomida (n=64), un grupo control pareado por enfermedad sin exposición a leflunomida durante el embarazo (n=108), o un grupo comparativo de mujeres embarazadas sanas (n=78). La exposición inadvertida a leflunomida durante el primer trimestre de embarazo, seguido de la descontinuación del medicamento y lavado del medicamento con colestiramina resultó en defectos estructurales mayores en 5.4% de los nacidos vivos comparado con alguno de los dos grupos de comparación (4.2% en el grupo pareado por enfermedad y 4.2% en mujeres embarazadas sanas).
Los resultados de este estudio, que fue descontinuado prematuramente debido a una disminución del reclutamiento, no modifican la contraindicación del uso de leflunomida durante el embarazo. Es importante recalcar que el estudio no investigó los posibles riesgos del uso de leflunomida durante el periodo embrionario completo, dado que todas las mujeres en el grupo expuesto a leflunomida descontinuaron la medicación al tener conocimiento del embarazo, casi todas tuvieron por lo menos un curso de tratamiento de eliminación del medicamento y la gran mayoría no estuvieron expuestas al medicamento luego de la tercera semana de gestación.
Estudios durante el postmercadeo: Un estudio multicéntrico, aleatorizado, controlado determinó la eficacia clínica de leflunomida con dos regímenes posológicos (con o sin dosis de carga) en pacientes con AR temprana sin exposición previa a DMARDS (n=121), utilizando los criterios ACR20 luego de tres meses como medida de desenlace primaria. Durante los primeros 3 días (periodo inicial doble –ciego), dos grupos en paralelo recibieron 20 mg o 100 mg de leflunomida con el placebo respectivo. El periodo inicial fue seguido por un periodo abierto de mantenimiento de 3 meses, durante el cual ambos grupos recibieron leflunomida 20 mg por día. La eficacia de leflunomida fue comprobada, pero no se demostró un beneficio adicional en el grupo que recibió la dosis de carga. Al finalizar el estudio, la tasa de respuesta ACR 20 fue 58.5% en el grupo que recibió dosis de carga comparado con 77.8% en el grupo sin dosis de carga (p=0.025). Para las medidas de desenlace secundarias analizadas (ACR50, ACR70, DAS28), no se demostraron diferencias significativas entre los dos grupos de tratamiento con un límite de probabilidad de 0.05. Se observó una respuesta clínica durante el primer mes de tratamiento en más de la mitad de los sujetos participantes, pero no se observaron diferencias significativas a los 30 días entre los grupos de tratamiento para todas las medidas de eficacia. Los datos de seguridad obtenidos de ambos grupos de tratamiento fueron consistentes con el perfil de seguridad ya conocido para leflunomida. Sin embargo, se observó una tendencia al aumento de la incidencia de eventos adversos gastrointestinales y de elevación de enzimas hepáticas en pacientes que recibieron la dosis de carga de 100 mg de leflunomida.
INTERFERENCIA CON TEST DE LABORATORIO Y DIAGNOSTICO
Abuso y dependencia: Leflunomida no tiene potencial conocido de abuso o dependencia.
ADVERTENCIAS: El riesgo de malignidad, en particular de desórdenes linfoproliferativos, podría ser aumentado con el uso de algunos medicamentos inmunosupresores.
PRECAUCIONES
Generales: Debido a la vida media prolongada del metabolito activo de Leflunomida, A771726, pueden ocurrir reacciones adversas o persistir, aun después que la administración de Leflunomida ha sido descontinuada. (Ver sección Reacciones adversas).
Si ocurre una reacción adversa severa a Leflunomida, o si por cualquier otra razón A771726 necesita ser depurado rápidamente del organismo, colestiramina o carbón activado debe ser iniciado como se describe en la sección 12, y continuar ó repetir según necesidad clínica. En casos de sospecha de reacciones alérgicas/inmunológicas severas una administración prolongada de colestiramina o carbón activado puede ser necesaria para alcanzar un aclaramiento rápido y suficiente, ver sección Embarazo.
La co-administración de Leflunomida con teriflunomida no es recomendada, ya que Leflunomida es el compuesto original de Teriflunomida.
Hígado: Dado que el metabolito activo de Leflunomida, A771726, se liga altamente a las proteínas y es depurado vía metabolismo hepático y secreción biliar, y dado el posible riesgo de hepatotoxicidad, Leflunomida deberá ser usada con precaución en los pacientes con alteración de la función hepática. Leflunomida no es recomendada en pacientes con deterioro hepático significativo o con enfermedad hepática pre-existente.
ALT (SGPT) debe ser evaluada antes de iniciar el tratamiento y por lo menos en intervalos mensuales durante los primeros 6 meses de tratamiento, y después cada 6 – 8 semanas.
Los lineamientos para ajustar la dosis o suspenderla son basados en la severidad y persistencia de la elevación de la ALT y son las siguientes: Para elevaciones confirmadas de ALT entre 2 y 3 veces el límite superior normal, reducciones de la dosis de 20 mg/día a 10 mg/día pueden permitir la continuidad de la administración de Leflunomida, bajo vigilancia estrecha.
Si el incremento de ALT (SGPT) entre 2 y 3 veces el límite superior normal persiste o si se confirman elevaciones superiores a 3 veces el límite superior normal, Leflunomida debe ser descontinuada. Colestiramina o carbón activado deberá ser administrado para disminuir más rápidamente los niveles de A771726.
Se han reportado casos raros de daño hepático severo, en casos aislados con desenlace fatal, durante el tratamiento con Leflunomida. La mayoría de éstos dentro de los primeros seis meses de tratamiento. Sin embargo una relación causal con Leflunomida no ha sido establecida y múltiples factores de confusión han estado presentes en la mayoría de los casos, se considera esencial que las recomendaciones de monitoreo sean seguidas estrictamente (ver arriba).
Sistemas hematopoyético e inmune: En pacientes con anemia pre-existente, leucopenia y/o trombocitopenia, así como en pacientes con alteraciones en la función de la médula ósea o aquellos con riesgo de supresión medular, el riesgo de desarrollo de reacciones hematológicas esta incrementado (ver sección Interacciones).
Un cuadro hemático completo, incluyendo un recuento diferencial de células blancas y plaquetas, deberá realizarse antes de iniciar la terapia con Leflunomida, así como mensualmente durante los primeros 6 meses de tratamiento y cada 6-8 semanas posteriormente.
Monitoreo hematológico frecuente (conteo completo de células sanguíneas, incluyendo conteo diferencial de glóbulos blancos y conteo de plaquetas) deberá ser realizado en
— Pacientes con tratamiento reciente o concomitante con medicamentos inmunosupresores o hematotóxicos y cuando el tratamiento con Leflunomida sea seguido por tales sustancias, sin un periodo previo de lavado.
— Pacientes con historia de anormalidades hematológicas relevantes.
— Pacientes con anormalidades hematológicas relevantes al momento de inicio de la terapia, debidas a causas diferentes a enfermedad artrítica.
Ver: Precauciones/General para las acciones que deben tomarse en caso de reacciones hematológicas severas.
Debido al potencial de inmunosupresión, aunque no hay experiencia clínica, la leflunomida no es recomendada en pacientes con
— Inmunodeficiencia severa (p. ej., SIDA).
— Deterioro significativo de la función de la médula ósea.
— Infecciones serias.
Infecciones: Medicamentos como Leflunomida, que tienen potencial inmunosupresor pueden hacer que los pacientes sean más susceptibles a infecciones, incluyendo infecciones oportunistas (ver sección Reacciones adversas). Las infecciones pueden ser más severas en naturaleza y pueden incluso requerir un tratamiento temprano y vigoroso.
En el evento de que una infección seria ocurra, puede ser necesario suspender el tratamiento con Leflunomida y administrar un procedimiento de lavado, tal como se describe en la sección Sobredosis.
Antes de iniciar el tratamiento, todos los pacientes deben ser evaluados para detectar tuberculosis activa ó inactiva (“latente”), siguiendo las recomendaciones locales. Los pacientes con historia de tuberculosis deben ser cuidadosamente monitoreados, por la posibilidad de reactivación de la infección.
— Respiratorio
Durante el tratamiento con Leflunomida raramente ha sido reportada enfermedad pulmonar intersticial (ver sección Reacciones adversas). El riesgo de su ocurrencia está aumentado en pacientes con historia de enfermedad intersticial pulmonar. La enfermedad pulmonar intersticial es un desorden potencialmente fatal, que puede ocurrir agudamente durante la terapia. Síntomas pulmonares como tos, disnea, pueden ser una razón para descontinuar la terapia y realizar una adecuada investigación posterior.
• Neuropatía periférica
Se han reportado casos de neuropatía periférica en pacientes que recibieron Leflunomida. La gran mayoría de los pacientes se recuperaron luego de la descontinuación de Leflunomida, pero algunos pacientes presentaron persistencia de los síntomas. Ser mayor de 60 años, la administración de medicamentos neurotóxicos concomitante y diabetes, pueden aumentar el riesgo de presentar neuropatía periférica. Si un paciente que esté tomando Leflunomida desarrolla neuropatía periférica, se debe considerar la descontinuación del tratamiento con Leflunomida y realizar el procedimiento de eliminación descrito en la sección Embarazo.
• Deterioro renal
En el momento no existe experiencia suficiente disponible para hacer recomendaciones específicas sobre la dosificación en los pacientes con alteración de la función renal. Debe tenerse precaución cuando se administre Leflunomida en esta población. Deberá considerarse que el metabolito activo de Leflunomida, A771726, se liga en forma importante a las proteínas.
• Reacciones en piel
Casos de síndrome de Stevens-Johnson, necrólisis epidérmica tóxica y la reacción a medicamentos, con eosinofilia y síntomas sistémicos (DRESS) han sido reportados en pacientes tratados con Leflunomida (ver sección Reacciones adversas). Si un paciente que toma Leflunomida desarrolla cualquiera de estas enfermedades de la piel, el tratamiento debe ser suspendido y los procedimientos de lavado se deben iniciar inmediatamente (ver sección Sobredosis).
• Presión sanguínea
La presión sanguínea debe ser revisada antes de iniciar el tratamiento y periódicamente durante el tratamiento.
• Uso en hombres
La información disponible no sugiere que la Leflunomida pueda estar asociada con un aumento del riesgo de toxicidad fetal-mediada por el hombre. Sin embargo, estudios en animales para evaluar este riesgo específico no han sido realizados. Para minimizar cualquier posible riesgo, los hombres que deseen concebir un hijo, deberán considerar la suspensión de Leflunomida y seguir el proceso de eliminación del fármaco descrito en la sección Embarazo.
ADMINISTRACIÓN: Las tabletas de ARAVA® deben ser deglutidas enteras, con suficiente líquido.
DOSIFICACIÓN Y ADMINISTRACIÓN: El tratamiento con leflunomida para la Artritis Reumatoidea (AR) deberá ser iniciada por médicos experimentados en el tratamiento de enfermedades reumatológicas.
Para recomendaciones de monitoreo ver sección Precauciones.
La terapia con la Leflunomida en artritis reumatoidea se inicia, usualmente, con una dosis de carga de 100 mg una vez al día durante 3 días. La omisión de la dosis de carga puede disminuir el riesgo de eventos adversos. Para información adicional relacionada con el uso de la dosis de carga de leflunomida en pacientes con artritis reumatoidea, ver sección Farmacodinamia. La dosis de mantenimiento recomendada es 20 mg de leflunomida una vez al día.
En caso que 20 mg sean pobremente tolerados, la dosis puede reducirse a 10 mg una vez al día.
La terapia con leflunomida para la Artritis Psoriásica (PsA) se inicia también con una dosis inicial de 100 mg una vez al día por 3 días. La dosis de mantenimiento es de 20 mg de leflunomida una vez al día.
El efecto del tratamiento puede evidenciarse después de 4 semanas y puede haber mejoría posterior después de 4 a 6 meses después de iniciado el tratamiento.
Usualmente ARAVA® se administra a largo plazo.
POBLACIONES ESPECIALES
Pacientes pediátricos: No se recomienda la administración de leflunomida en pacientes menores de 18 años de edad, pues no ha sido estudiada en este grupo.
Pacientes ancianos: No se requiere ajuste de dosis en ancianos (pacientes mayores de 65 años de edad).
Deterioro hepático
Ver: Sección Precauciones.
Deterioro renal:
Ver: Sección Precauciones.
SOBREDOSIS
Signos y síntomas: Ha habido reportes de sobredosis crónica en pacientes que tomaron dosis diarias de ARAVA® de hasta cinco veces la dosis recomendada diaria y reportes de sobredosis aguda en adultos o niños. No se reportaron eventos adversos en la mayoría de los casos de sobredosificación. Los eventos adversos fueron consistentes con el perfil de seguridad de ARAVA® (ver sección Reacciones adversas). Los eventos adversos más frecuentemente observados fueron diarrea, dolor abdominal, leucopenia, anemia y elevación de las pruebas de función hepática.
Manejo: En el evento de sobredosis relevante o toxicidad, la colestiramina o carbón activado están recomendados para acelerar la eliminación. La colestiramina administrada por vía oral en una dosis de 8 g tres veces al día, durante 24 horas en tres voluntarios sanos disminuyó los niveles plasmáticos de A771726 en aproximadamente 40% en 24 horas y en 49 - 65% en 48 horas.
La administración de carbón activado (polvo convertido en suspensión) oral o por sonda nasogástrica (50 g cada 6 horas durante 24 horas) ha demostrado reducir las concentraciones en plasma del metabolito activo A771726 en 37% en 24 horas y en 48% en 48 horas.
Estos procedimientos de lavado pueden ser repetidos si es clínicamente necesario.
Estudios con hemodiálisis y CAPD (diálisis peritoneal ambulatoria crónica) indican que A771726, el metabolito primario de Leflunomida no es dializable.
DESCRIPCIÓN
Moléculas activas / Ingredientes Activos: Leflunomida.
Clase terapéutica o farmacológica: Antirreumático, inmunomodulador, Código ATC L04AA13
PRESENTACIONES: ARAVA® 20 mg, frasco por 30 tabletas recubiertas (Reg. San. INVIMA 2009 M-013233-R1).
ARAVA® 100 mg, caja blíster por 3 tabletas recubiertas (Reg. San. INVIMA 2009M-013232 R1).
CCDS V20
SANOFI-AVENTIS DE COLOMBIA S. A.
Transversal 23 No. 97-73, Pisos 8 y 9
Teléfono: 6214400, Fax: 7444237
Bogotá, D.C., Colombia
CONDICIONES DE ALMACENAMIENTO Y VIDA DE ALMACENAJE: Almacenar a temperatura inferior a 30 °C.
Tiempo de vida útil:
— ARAVA® 20 mg: 2 años.
— ARAVA® 100 mg: 3 años.

